Biosecurity working example
Abagael Sykes, Gustavo Machado
2022-01-12
Source:vignettes/Vignette_biosecurity.Rmd
Vignette_biosecurity.RmdGeneral introduction
MrIML is a R package allows users to generate and interpret multi-response models (i.e., joint species distribution models) leveraging advances in data science and machine learning. MrIML couples the tidymodel infrastructure developed by Max Kuhn and colleagues with model agnostic interpretable machine learning tools to gain insights into multiple response data such as. As such MrIML is flexible and easily extendable allowing users to construct everything from simple linear models to tree-based methods for each response using the same syntax and same way to compare predictive performance.In this vignette we will guide you through how to apply the MrIML framework to identify and benchmark biosecurity practices in veterinary disease. This working example is built upon a classification framework. More information about this can be found in the “Classification working example”.
Lets load that data
This is a synthesized dataset to simulate PRRSV infection, biosecurity practices and farm demographics on swine farms across the united states. Please note that the data included in this package is simulated, and is not a reflection of any real farm, company or state.
#other packages needed
library(mrIML);library(parsnip);
library(tidymodels); library(workflows);
library(tidyverse); library(flashlight);
library(devtools); library(DALEX);
library(plyr);library(vip);
library(ggsci); library(ggrepel);
library(breakDown); library(future.apply);
library(janitor)
set.seed(130) #set the seed to ensure consistency
data <- as.data.frame(mrIML::biosecurity_vignette_data)Defining your model engine
This is a random forest model, as used in the classification example. However there are more models available within MrIML and can be found here.
#split predictor variables and outcome
Y <- as.data.frame(data %>% select(1))
X <- data %>% select(-c(1, 44, 45)) #remove class, group and ID
model1 <- #model used to generate yhat
#specify that the model is a random forest
rand_forest(trees = 100, mtry = tune(), min_n = tune(), mode = "classification") %>%
#select the engine/package that underlies the model
set_engine("ranger", importance = c("impurity","impurity_corrected")) %>%
#must be set to classification for classification problems
set_mode("classification")###Parallel processing
MrIML provides uses the flexible future apply functionality to set up multi-core processing. In the example below, we set up a cluster using 4 cores. If you don’t set up a cluster, the default settings will be used and the analysis will run sequentially.
# detectCores() #check how many cores you have available. We suggest keeping one core free for internet browsing etc.
cl <- parallel::makeCluster(4)
plan(cluster, workers=cl)Running the analyis
Now we can train and test our model.
yhats <- mrIMLpredicts(X=X, #features/predictors
Y=Y, #response data
Model=model1, #specify your model
balance_data='no', #chose how to balance your data
mode='classification', #chose your mode (classification versus regression)
seed = 120) #set seedWe can then assess our model performance using a number of performance metrics including area under the curve (AUC), sensitivity, specificity and Matthews correlation coefficient (MCC).
ModelPerf <- mrIMLperformance(yhats=yhats, Model=model1, Y=Y, mode='classification')
ModelPerf[[1]] #predictive performance for individual responses
#> response model_name roc_AUC mcc sensitivity
#> 1 Class rand_forest 0.658615136876006 0.269515754526925 0.740740740740741
#> specificity prevalence
#> 1 0.521739130434783 0.5
ModelPerf[[2]]#overall predictive performance. r2 for regression and MCC for classification
#> [1] 0.6586151Benchmarking: global importance
Here we can look at the variable importance. This is the dependence between our outcome and biosecurity variables.
#calculate variable importance
VI <- mrVip(yhats=yhats, X=X)
#plot cumulative variable importance
plot_vi_biosecurity(VI=VI, Y=Y, X=X,
modelPerf=ModelPerf,
cutoff= 0.6,
plot.pca='no') #Follow instructions in console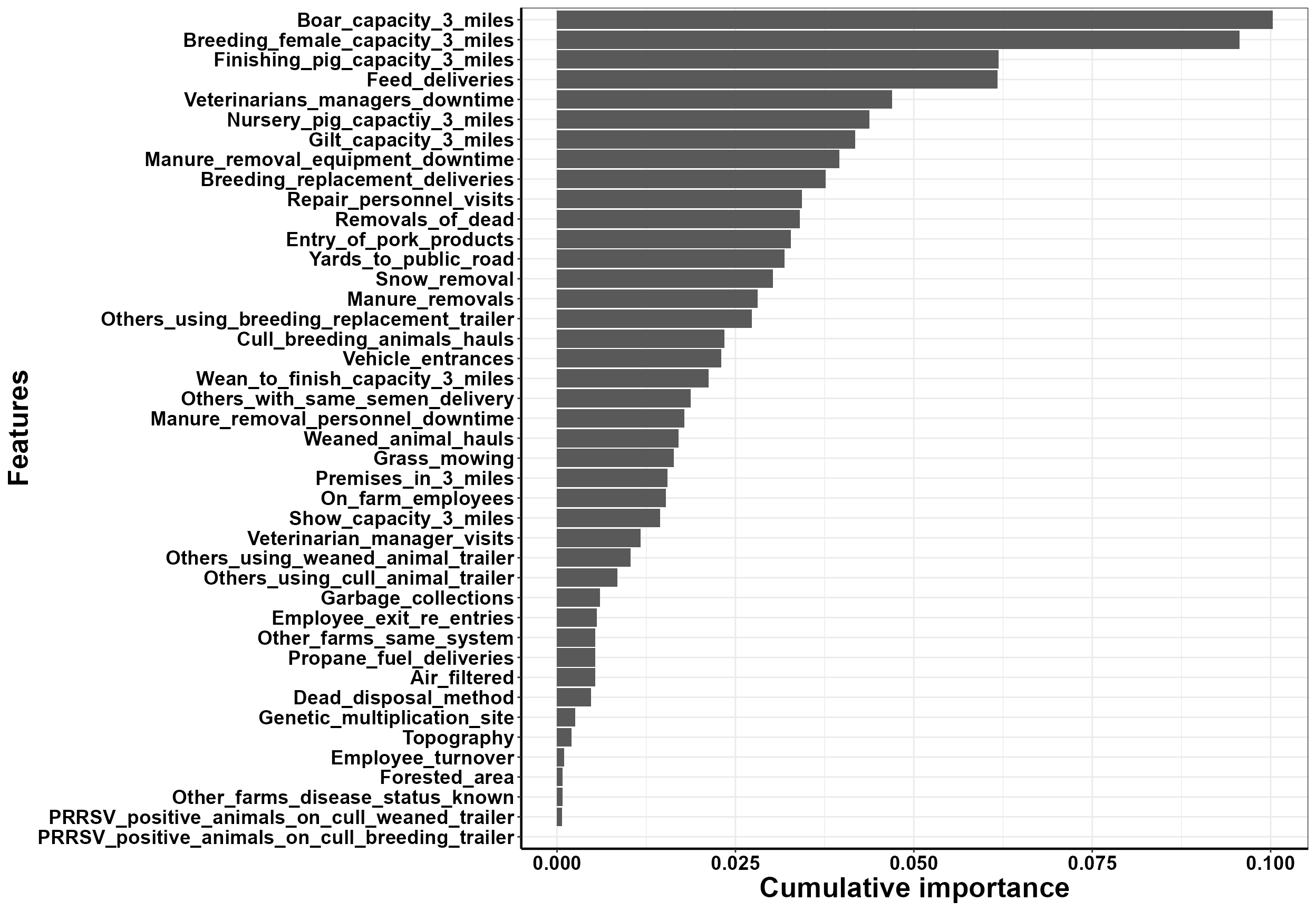
#> Press [enter] to plot individual variable importance summaries
#> [1] ""We can explore further and assess the partial dependence of each variable. Here we isolate the dependence of one variable and visualize how this dependence changes over different observed values.
#create a flashlight object
fl <- mrFlashlight(yhats=yhats, X=X, Y=Y,
response="single",
index=1,
mode='classification')
#plot partial dependence profiles
plot(light_profile(fl, v = "Premises_in_3_miles")) 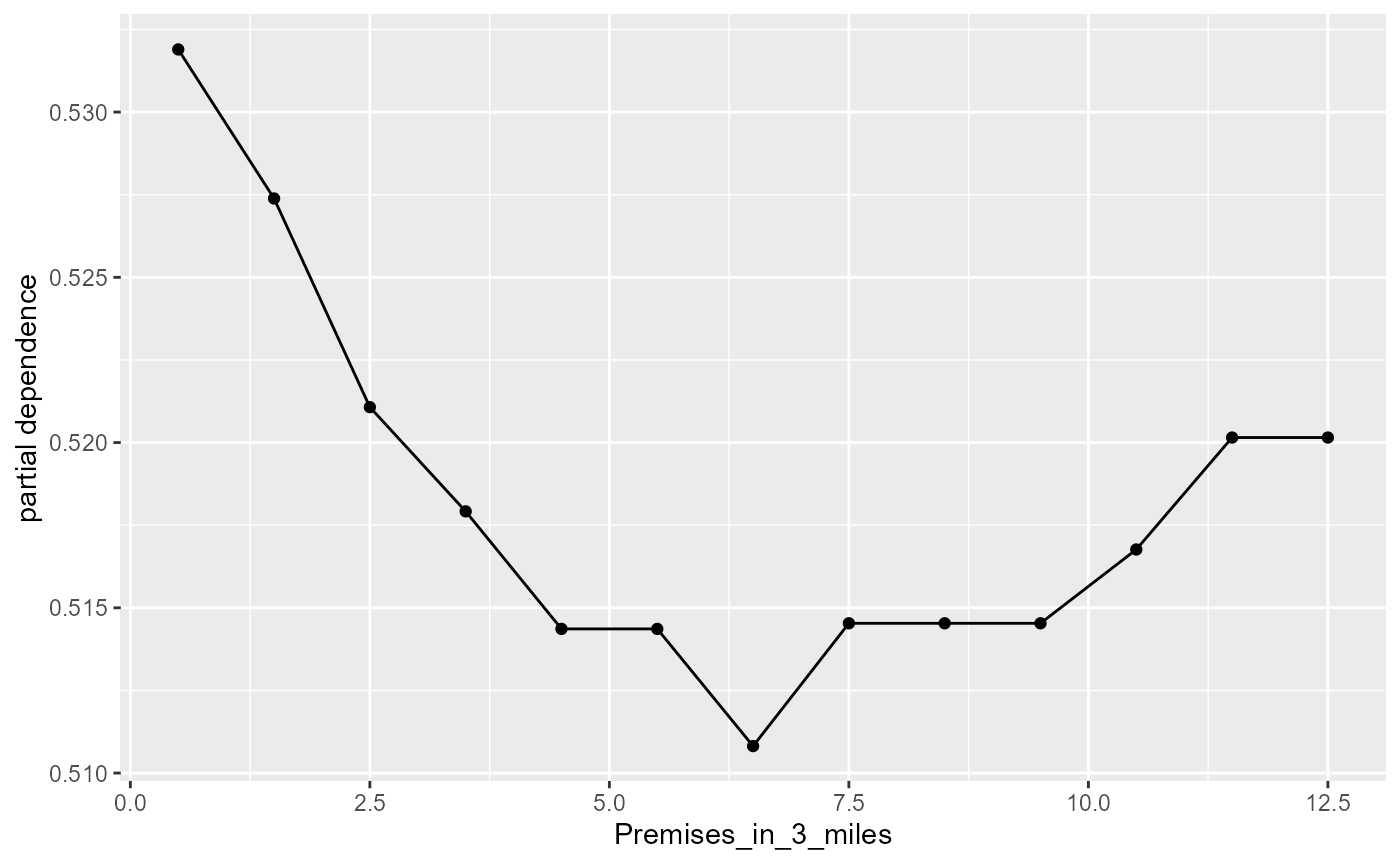
Benchmarking: predicted risk
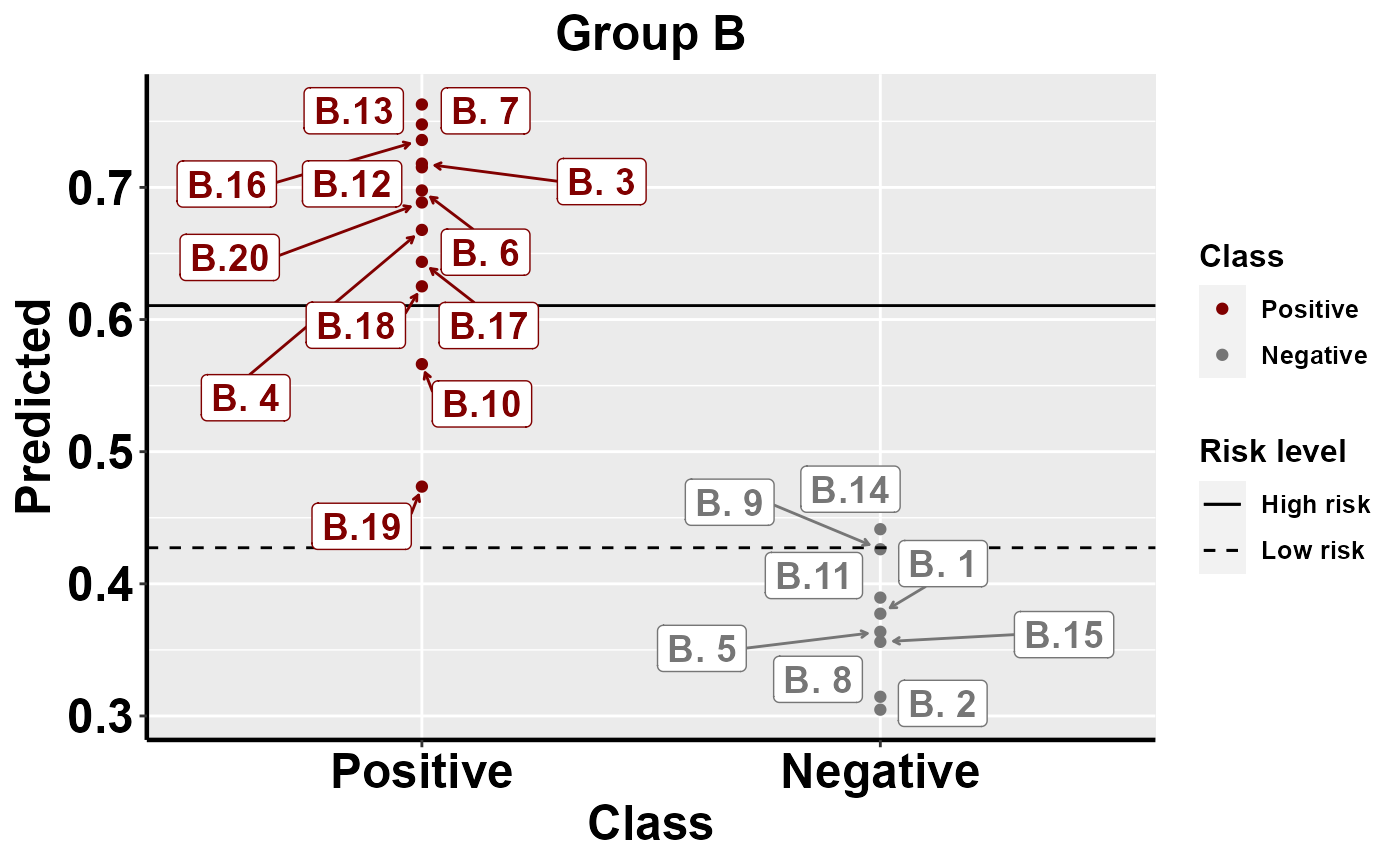
The following function allows you to visualize and compare predicted risk among and within groups
#apply the trained model to the entire data set to provide risk of predicted outbreak
fit_bio <- pull_workflow_fit(yhats[[1]]$mod1_k)
preds_pos <- predict(fit_bio, data, "prob")
data$Predicted <- preds_pos$.pred_1
#process data ready for the function
data$Class <- as.factor(data$Class)
data<-data%>%
mutate(Class = revalue(Class,
c("1" = "Positive", "0" = "Negative")))
data$Class <- relevel(data$Class, "Positive")
data1 <- data
#plot among group comparison of predicted risk
mrBenchmark(data = "data1", Y = "Class", pred = "Predicted", group = "Group", type = "external") 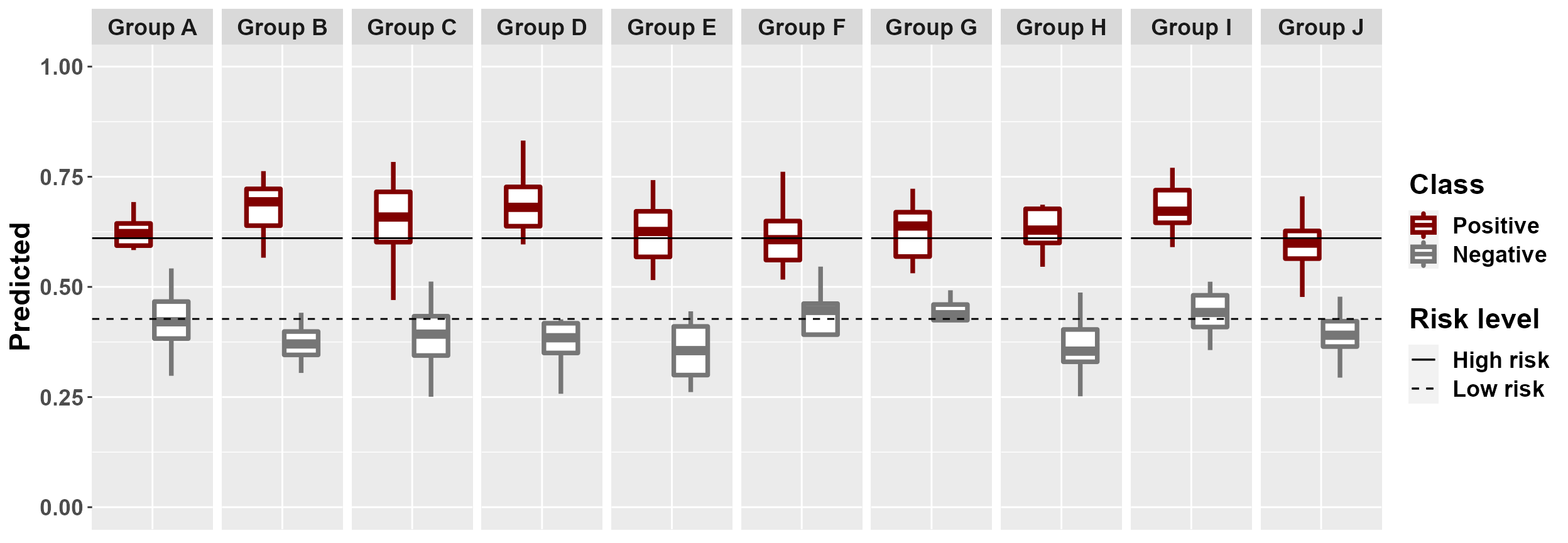
#plot within group comparison of predicted risk
mrBenchmark(data = "data1", Y = "Class", pred = "Predicted", group = "Group", label_by = "ID", type = "internal") 

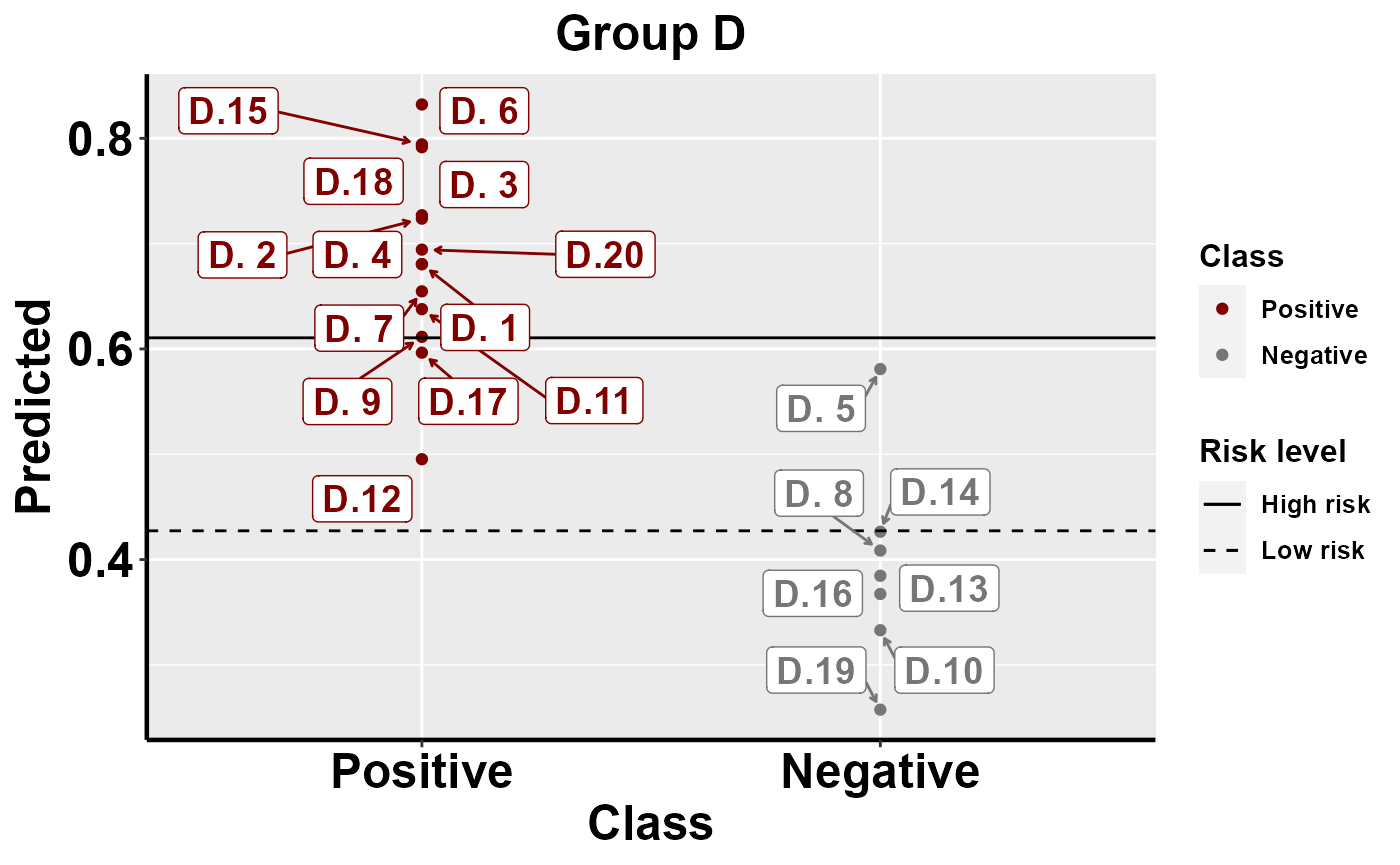

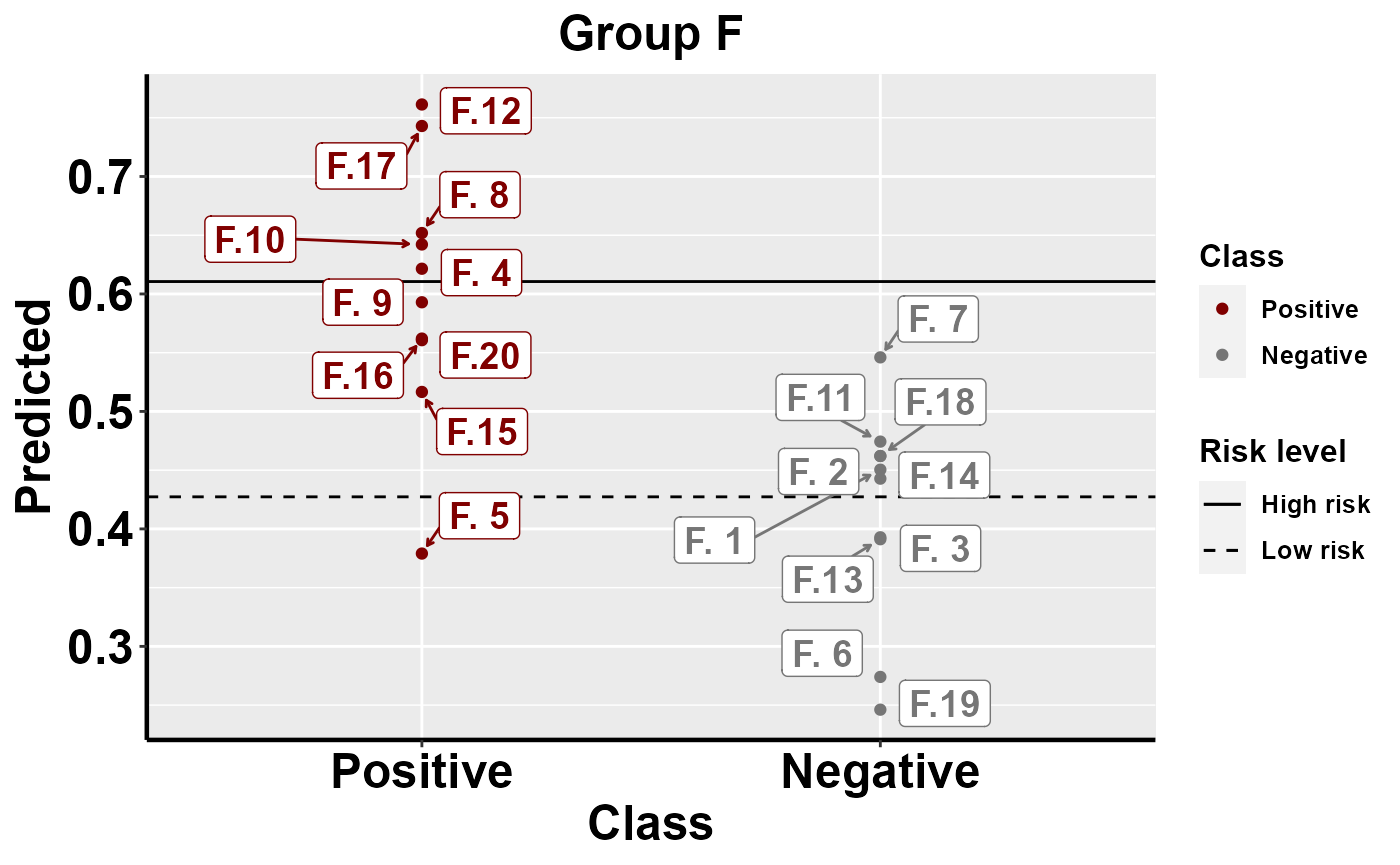
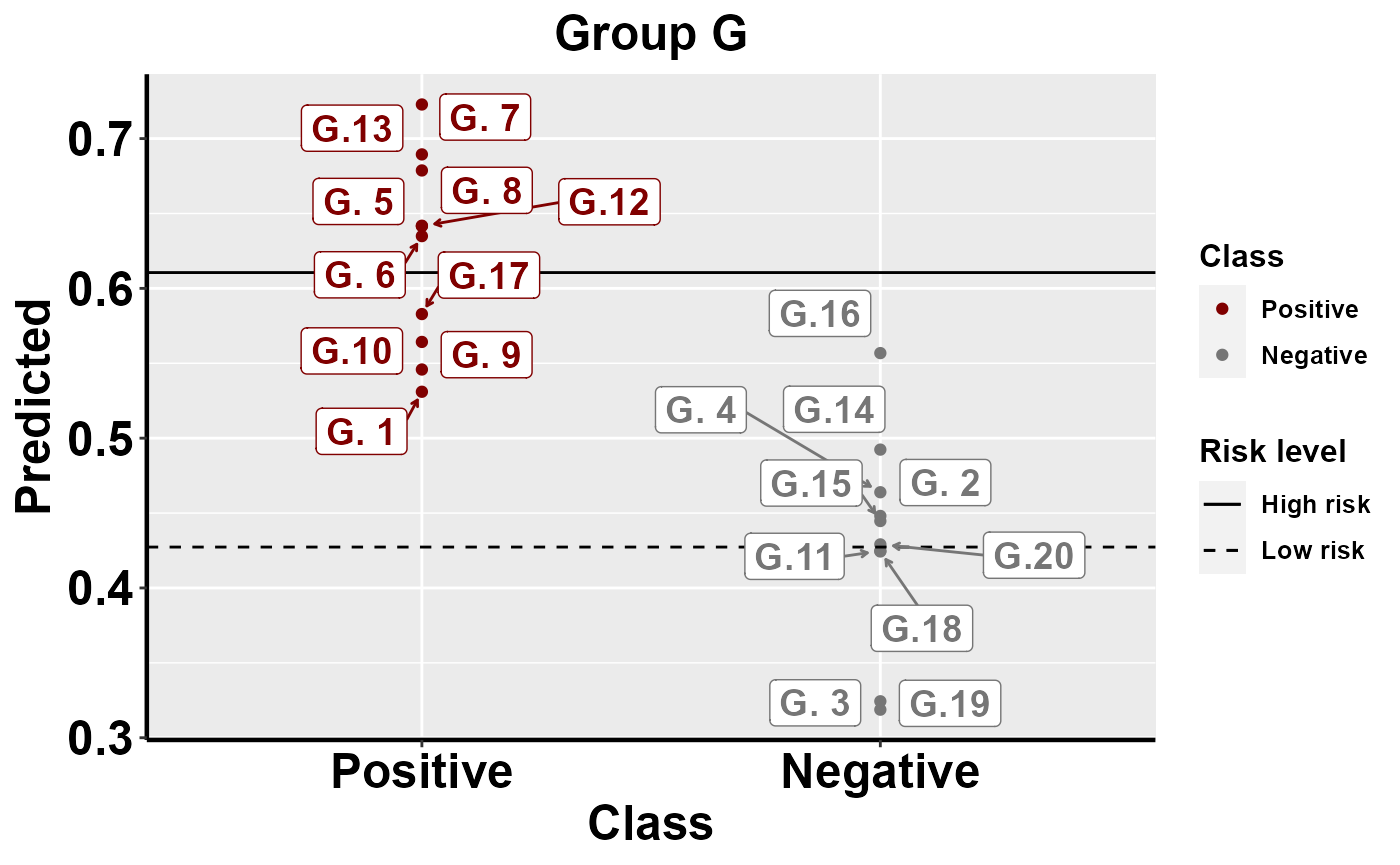
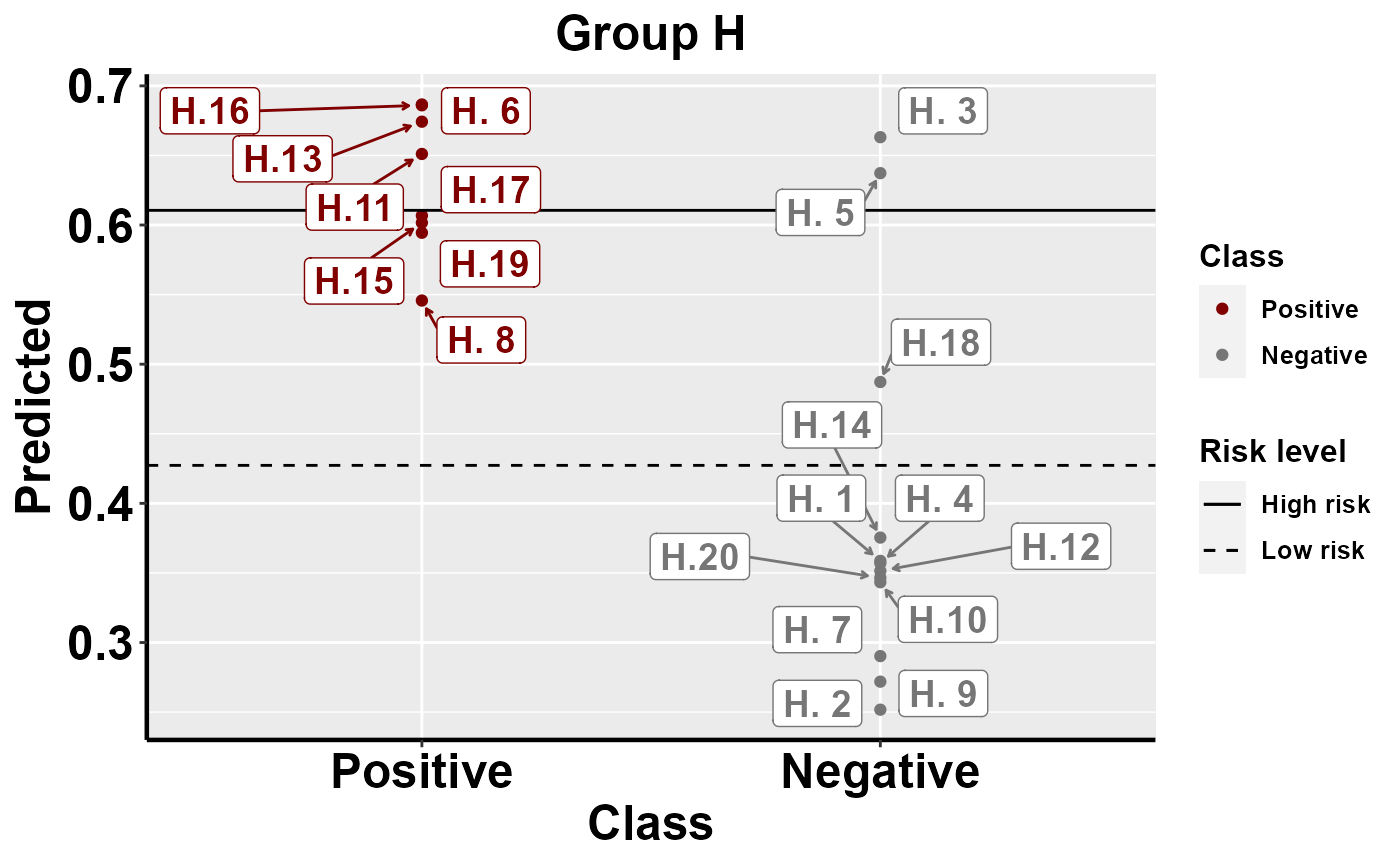
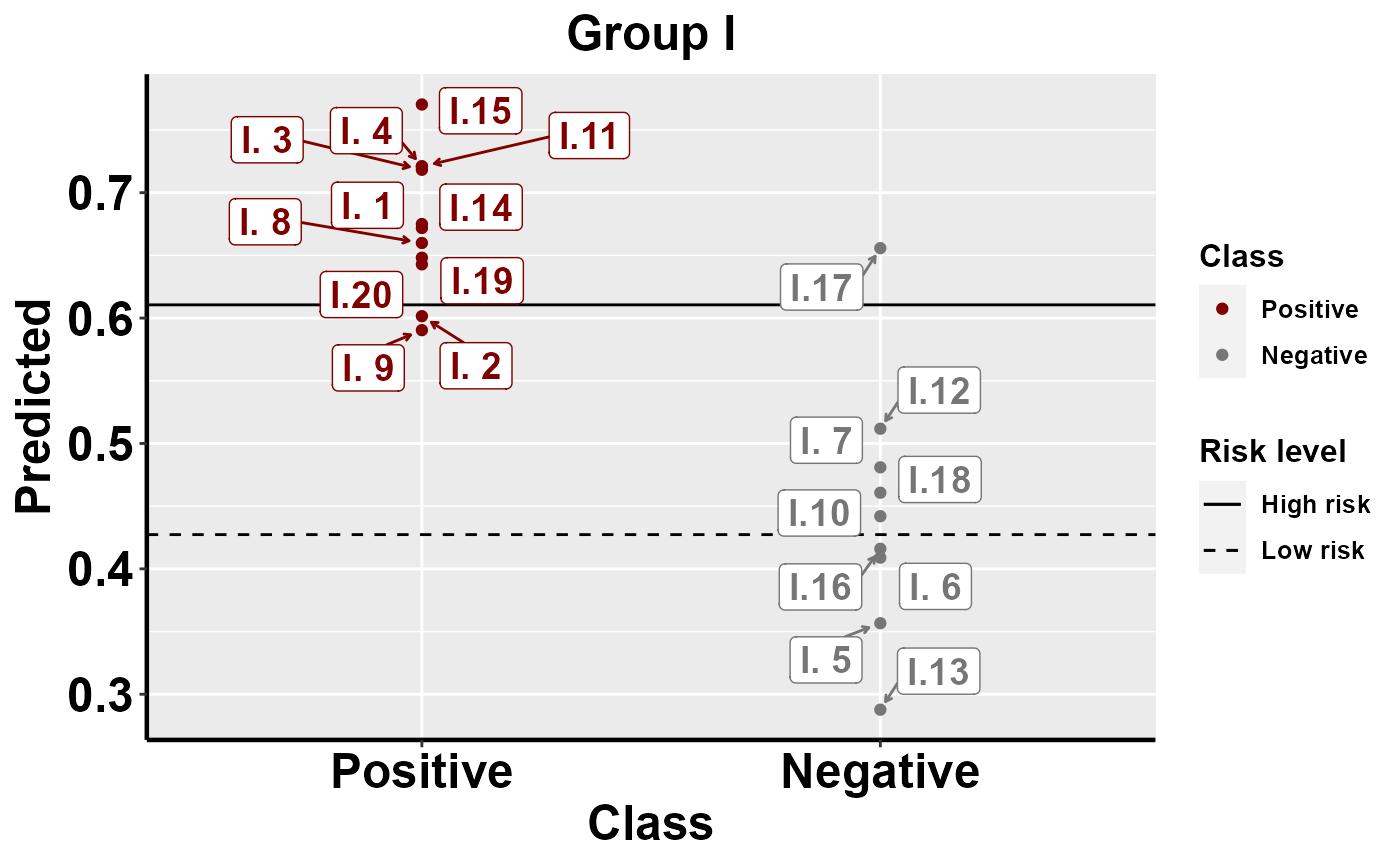
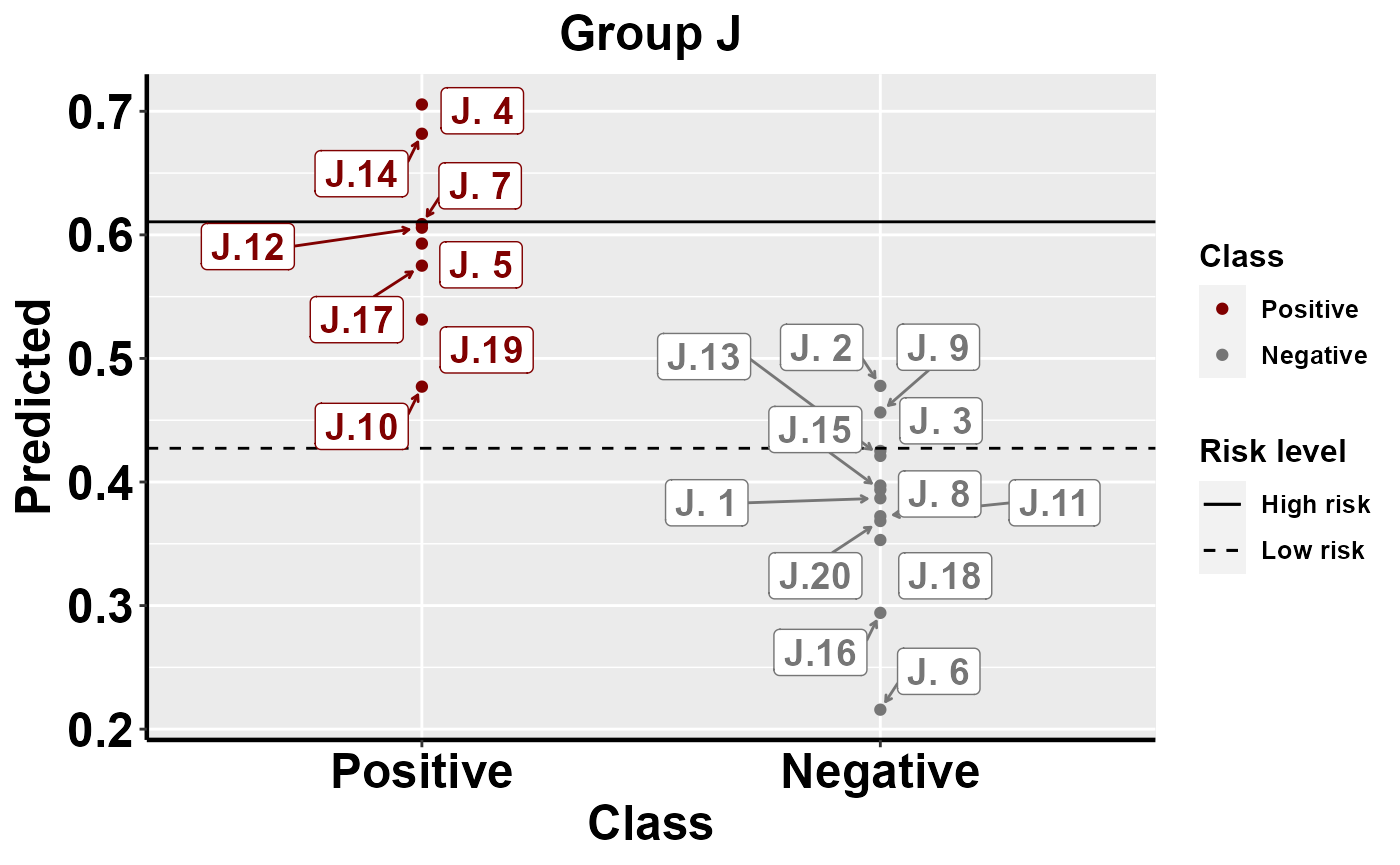
Benchmarking: local importance
To investigate and interpret the contribution of variables at an individual level, we must use a local explanation method. Here we implement a local breakDown explainer. mrLocalExplainer produces both aggregated and individual results of variable contribution. Variables with a phi > 0 contribute to an increase in predicted PRRSV outbreak risk, while variables with a phi < 0 contribute to a decrease in predicted outbreak risk.
#Use this function to implement the local explainer
data<-data%>%
mutate(Class = revalue(Class,
c("1" = "Positive", "0" = "Negative")))
data$Class <- relevel(data$Class, "Positive")
X1 <- X
mrLocalExplainer(X = X1, Model = yhats, Y = data$Class)
#> Preparation of a new explainer is initiated
#> -> model label : ranger ( default )
#> -> data : 200 rows 42 cols
#> -> target variable : not specified! ( WARNING )
#> -> predict function : yhat.ranger will be used ( default )
#> -> predicted values : No value for predict function target column. ( default )
#> -> model_info : package ranger , ver. 0.13.1 , task classification ( default )
#> -> model_info : Model info detected classification task but 'y' is a NULL . ( WARNING )
#> -> model_info : By deafult classification tasks supports only numercical 'y' parameter.
#> -> model_info : Consider changing to numerical vector with 0 and 1 values.
#> -> model_info : Otherwise I will not be able to calculate residuals or loss function.
#> -> predicted values : numerical, min = 0.160133 , mean = 0.5113279 , max = 0.8878627
#> -> residual function : difference between y and yhat ( default )
#> A new explainer has been created! 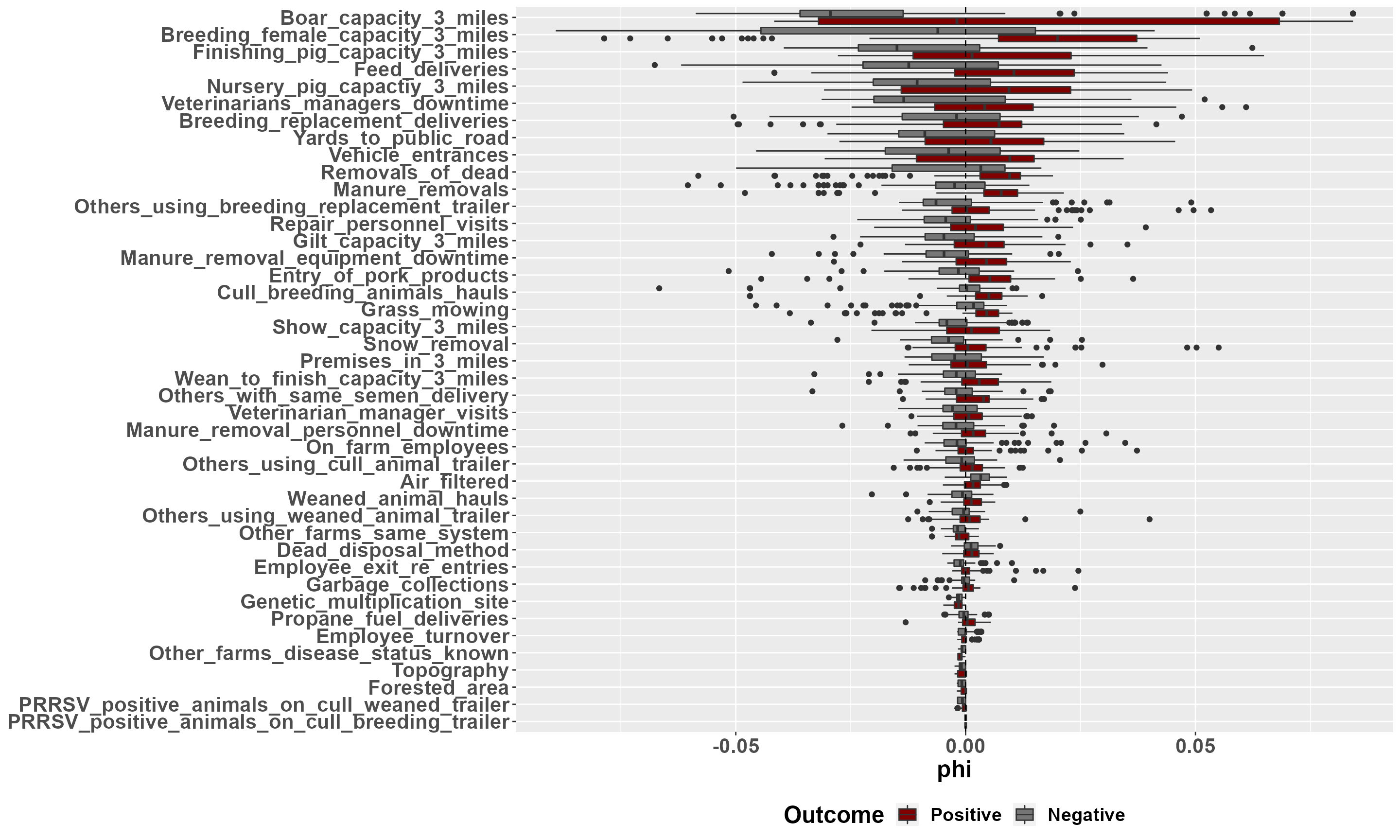
Individual plots are produced in the list object LE_indiv_plots. Each plot corresponds to a single farm. An example of a positive farm and a negative farm are shown here.
LE_indiv_plots[1]
#> [[1]]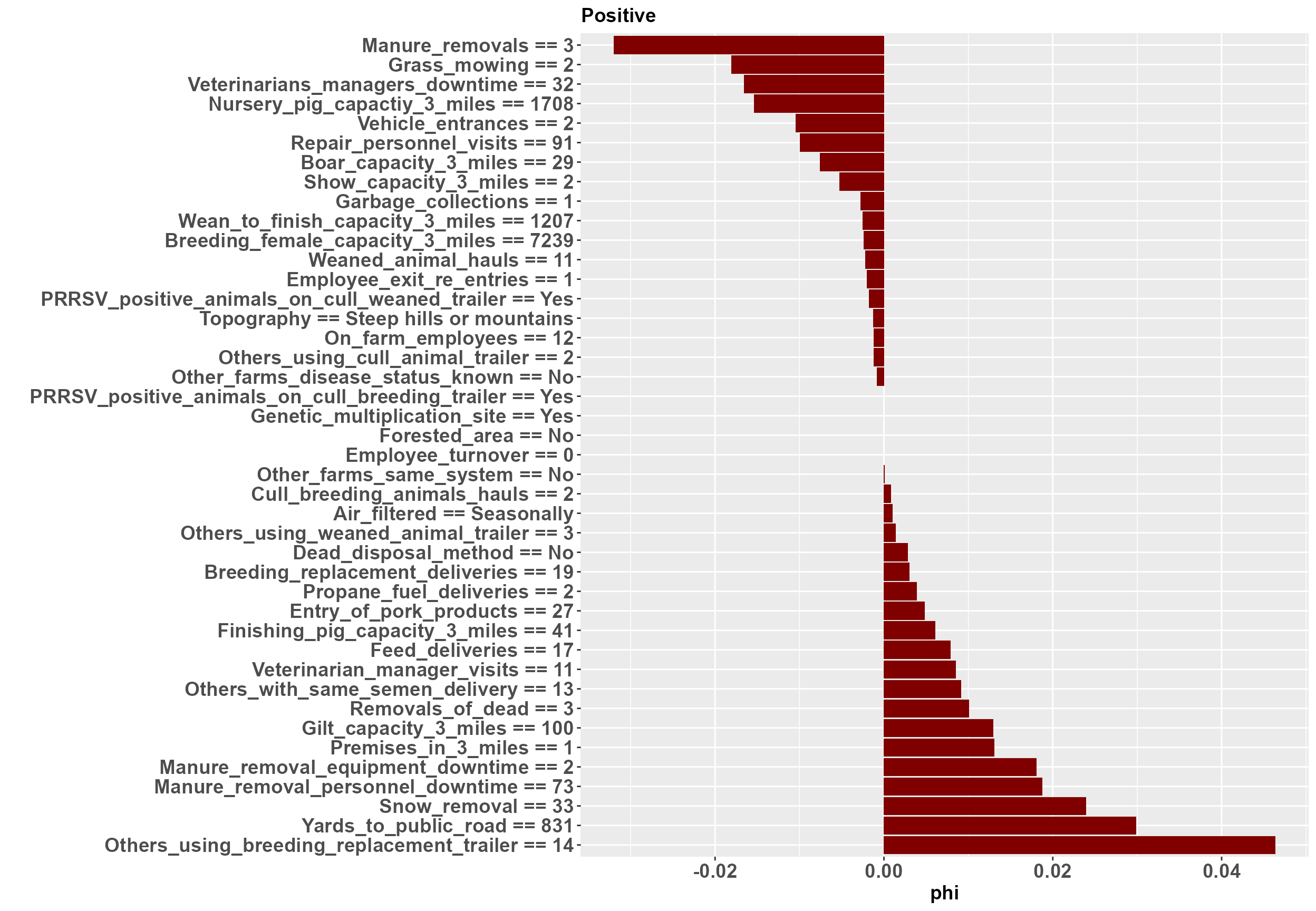
LE_indiv_plots[2]
#> [[1]]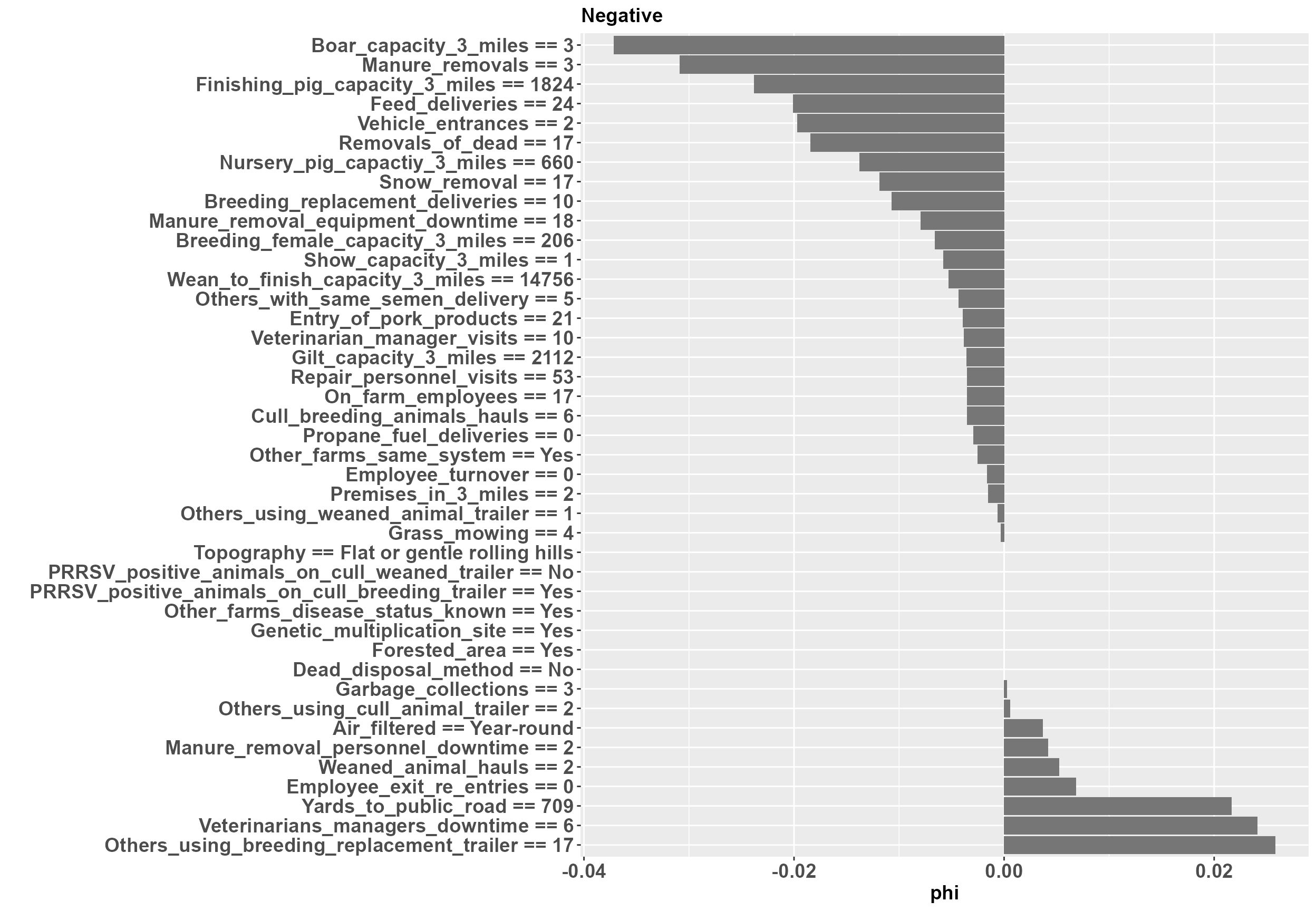 At the positive farm we can see that yards from the public road and garbage collection are contributing to an increased outbreak risk, while manure removals and repair visits are contributing to a decreased outbreak risk. Likewise, at the negative farm yards to the public road and and the downtime required for manure removal personnel are contributing to an increased outbreak risk, while feed deliveries and manure removals are contributing to a decreased outbreak risk.
At the positive farm we can see that yards from the public road and garbage collection are contributing to an increased outbreak risk, while manure removals and repair visits are contributing to a decreased outbreak risk. Likewise, at the negative farm yards to the public road and and the downtime required for manure removal personnel are contributing to an increased outbreak risk, while feed deliveries and manure removals are contributing to a decreased outbreak risk.
This session of the MrIML has been funded by Critical Agricultural Research and Extension 2019-68008-29910 from the USDA National Institute of Food and Agriculture.